1.Explanations of variants outputs
(1) Guidance for submission:
In the "Missense Mutation Pathogenicity Prediction" row, users can input multiple variants once. Notably, there are two format requirements, as below:
#CHROM POS REF ALT
1 1168196 C T
2 1481120 G T
3 9730718 A G
4 515710 G C
5 218471 A T
6 109787446 C T
7 195679 C T
8 1719221 A G
9 215012 C A
11 532671 G C
12 862859 C G
13 20567666 G C
14 20915433 C T
15 23086278 G C
16 136789 A G
16 138725 T A
16 138755 C T
16 139765 G A
16 139784 G C
17 436083 T C
19 620430 C T
20 398445 C G
20 400365 A G
21 16337059 G A
21 16339874 T C
22 17584454 G A
etc.
OR, users can upload "variants.vcf" file with the correct format.
(3) According to the variants information input by the user, the programs will calculate the characteristics of the variants, including:
Variant-level annotation features using Ensembl VEP v104 with critical pluing dbNSFP V4.1a;
We also applied other databases, such as ClinVar(202012); HGMD-PUBLIC(20204);dbSNP(v154);
Amino Acid-level embeddings. Two tools (ESM-1b and ProtT5-XL-U50) are adopted;
Genome-level annotation features, such as gnomAD_AFR_AF and ExAC_SAS_AF.
(4) Taking about 100 variants as one unit, we listed the time required for each part during the prediction procedure as below:
Ensembl VEP v104 with critical pluing dbNSFP V4.1a (need ~3 minutes);
Gererating protein sequence using API from web (need ~ 4 minutes);
ESM-1b amino acid-level embeddings (need ~ 5 minutes);
ProtT5-XL-U50 amino acid-level embeddings (need ~ 5 minutes);
Missense mutation pathogenicity prediction using ConsMM model (need ~ 2 minutes);
Missense mutation pathogenicity prediction using EvoIndMM model (need ~ 2 minutes).
Accordingly, users shouldn't wait for results on the waiting page. Once the prediction finished, the results link would send to your email address.
In view of the limitation of computing storage resources, the user's prediction results will be kept for 72 hours. Please download and save the result file when the prediction finished.
(2) Feature descriptions:
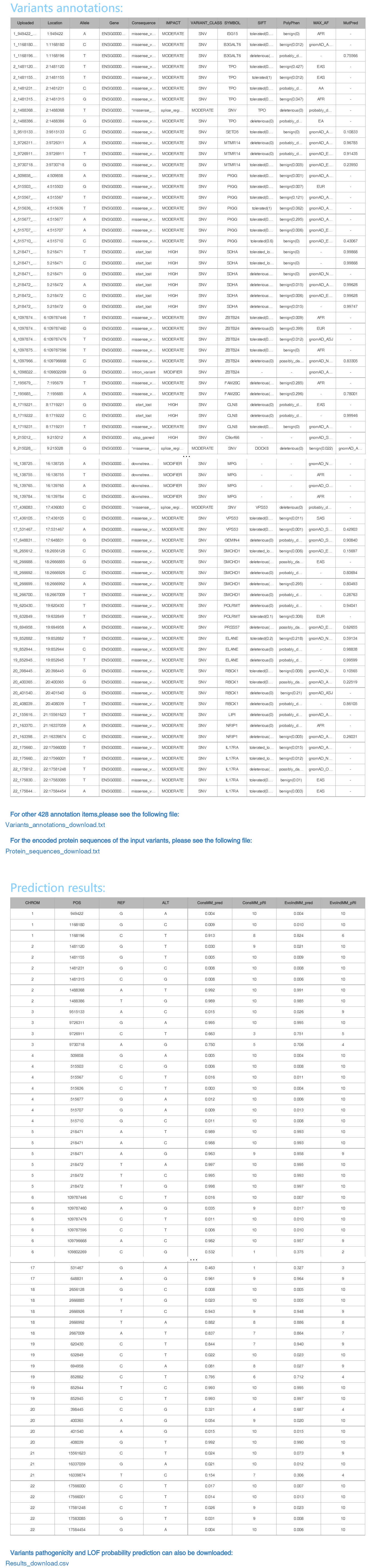
Using Ensembl VEP v104 with some plugins (such as dbNSFP4 verison 4.1a, dbSNP version 154, COSMIC version 92, etc), we can get multiple annotation information for input variants. Here, we listed some typical items as below:
For other feature descriptions,please see the supplementary material of this manuscript. OR,please see http://grch37.ensembl.org/info/docs/tools/vep/script/vep_plugins.html and https://sites.google.com/site/jpopgen/dbNSFP.
2.Example outputs
"Variants_annotations_download.txt": this file contains all 440 annotation items for the input variants.
"Protein_sequence_download.txt": this file contains the encoded protein sequences of the input variants.
"Results_download.csv": this file contains all variants pathogenicity probability prediction results, predicted by ConsMM and EvoIndMM models.
"ConsMM_pRI" and "EvoIndMM_pRI" are reliability Index of prediction score. Reliability Index formula is give below: RI = round(20*abs(P(patho)-0.5))