MMPatho: Interpretable Model with Multilevel Consensus and Evolutionary Information for Missense Mutation Pathogenic Prediction and Annotation
Abstract:
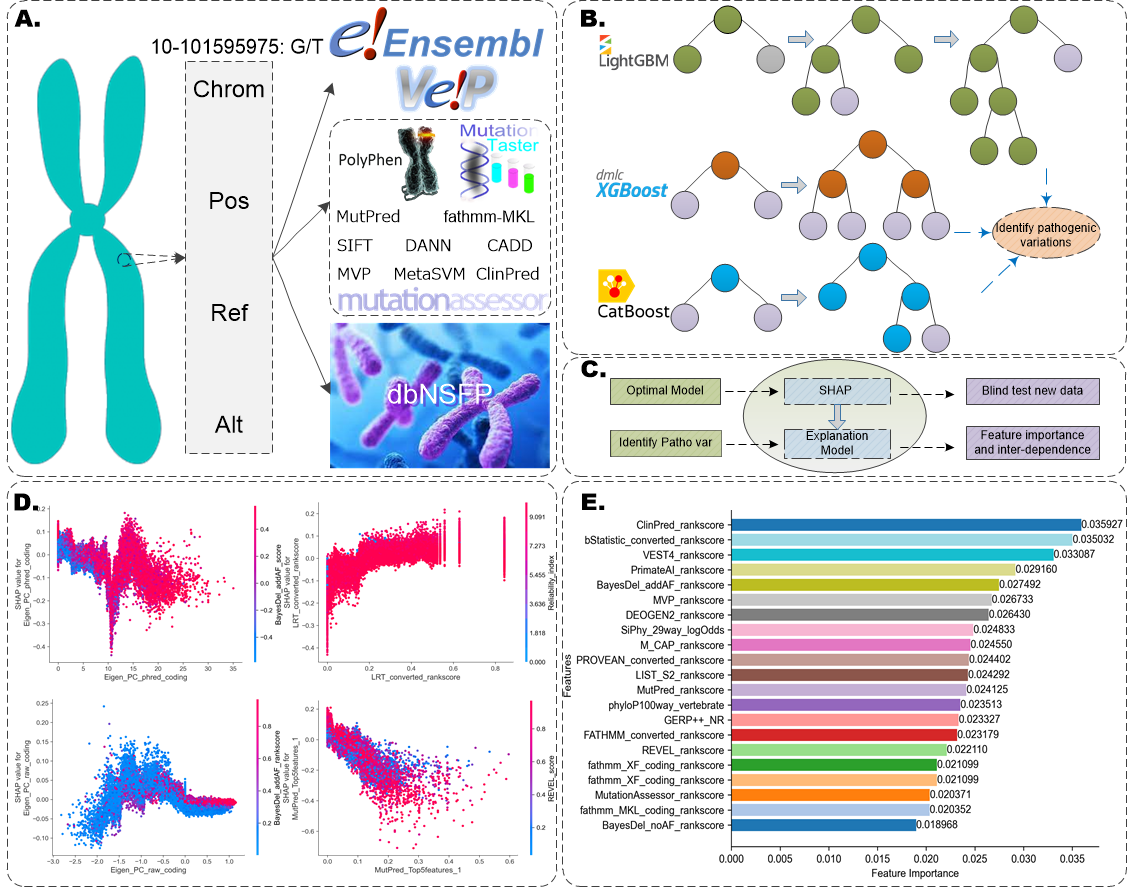
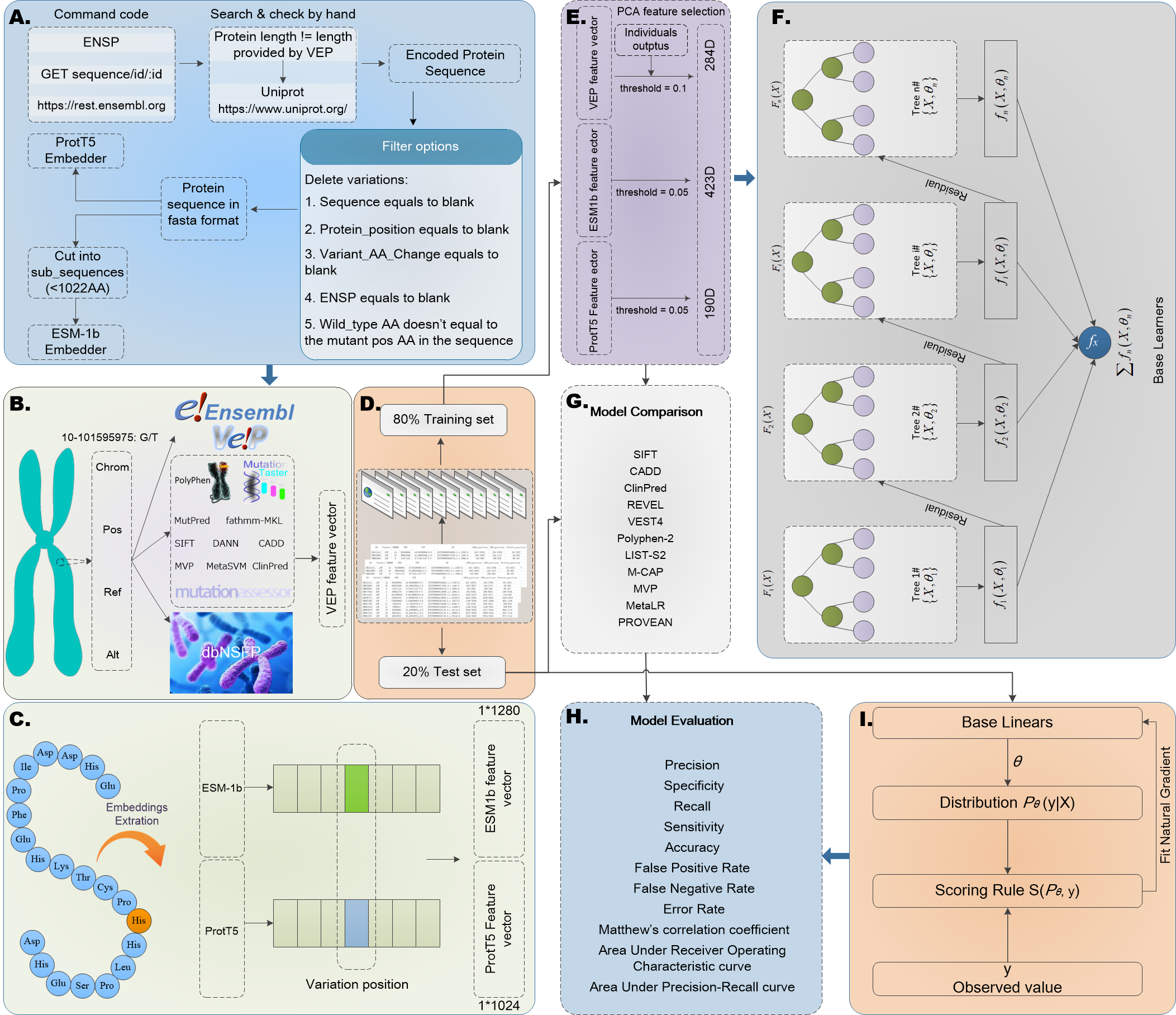
Comprehending the pathogenicity of missense mutation (MM) is crucial for elucidating genetic diseases, gene functions, and individual variations. In this work, we constructed a comprehensive large-scale non-redundant MM benchmark dataset built on the entire Ensembl database and a blind test set focused specifically on pathogenic GOF/LOF MM. For each mutation, we utilized Ensembl VEP v104 and dbNSFP v4.1a to obtain variant-level, amino acid-level, individuals' outputs, and genome-level features. Additionally, we adopted the ENSP identifier and Ensembl API to generate the encoded protein sequence, and then extracted ESM-1b and ProtTrans-T5 embeddings for each mutant site. Building upon these efforts, we developed an interpretable model group MMPatho (consisting of ConsMM and EvoIndMM). ConsMM utilizes individuals' outputs and the XGBoost algorithm with SHAP explanation analysis. Additionally, EvoIndMM examines whether incorporating evolutionary information from ESM-1b and ProtT5-XL-U50, large protein language embeddings, can further enhance the predictive capability. Through extensive comparative experiments, ConsMM and EvoIndMM exhibited impressive AUROC and AUPR values of 0.9856 and 0.9876, 0.9877 and 0.9919, respectively, on the blind test set, underscoring their efficacy in predicting MM pathogenicity.