MMPatho: Interpretable Model with Multilevel Consensus and Evolutionary Information for Missense Mutation Pathogenic Prediction and Annotation
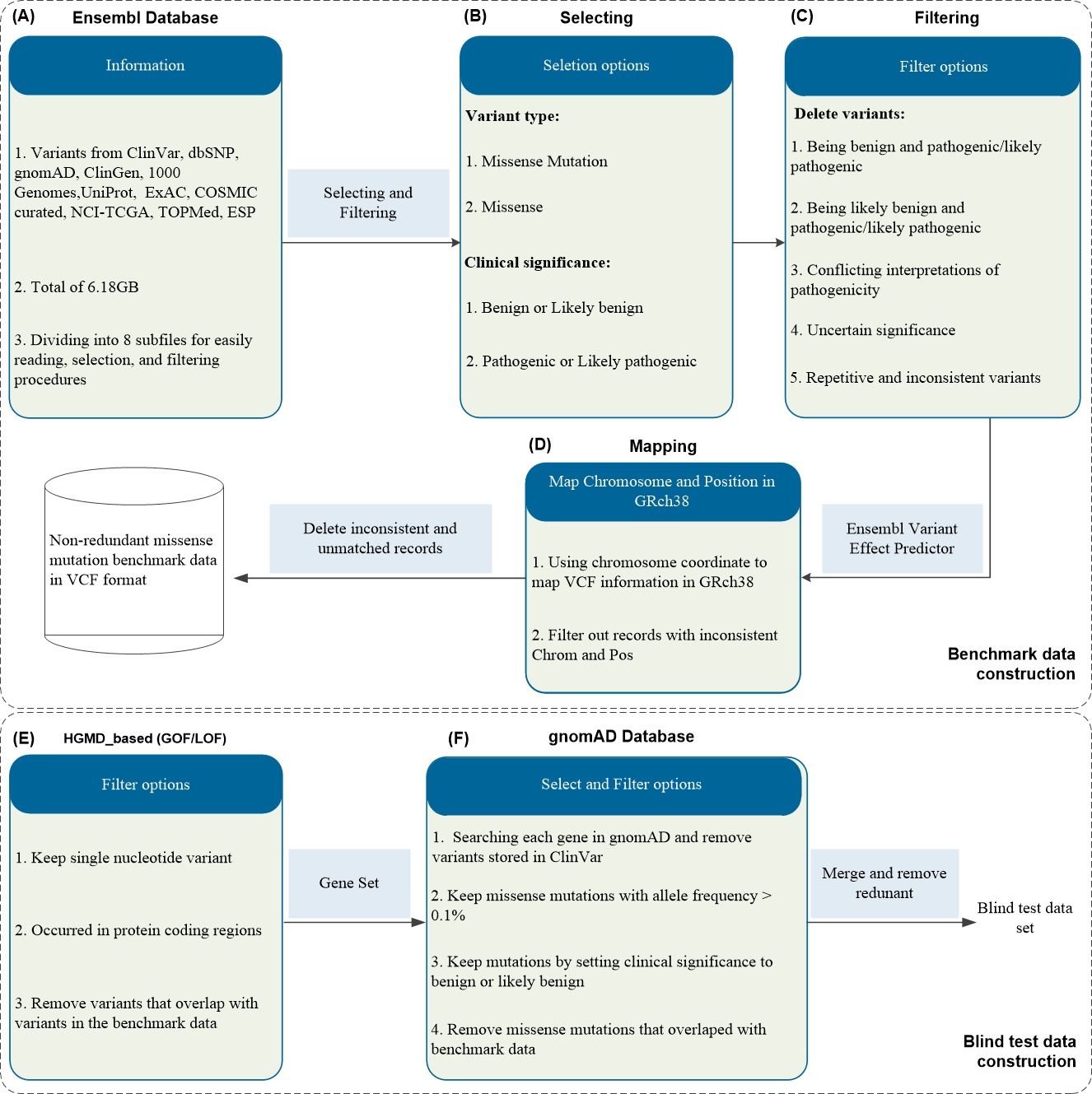
Figure 1. The overall workflow of benchmark dataset and blind test set construction.
- We downloaded the entire Ensembl database (6.18GB) from https://web.expasy.org/swissvar.html, which includes variants from various sources (e.g., ClinVar [1], dbSNP [2], gnomAD [3], 1000 Genomes [4], UniProt [5], ExAC [6], COSMIC [7]) (Figure 1A). To select the relevant MMs, we applied two filters: variant type (setting as MM or missense) and clinical significance (setting as benign/likely benign or pathogenic/likely pathogenic), getting 622,270 variants (Figure 1B). We then applied five additional filters to remove unwanted mutations: (1) those with conflicting interpretations of pathogenicity, (2) uncertain significance, (3) repetitive and inconsistent MMs from different sources, and (4) variants classified as benign and pathogenic/likely pathogenic, (5) variants being likely benign and pathogenic/likely pathogenic (obtaining 91,072 variants, Figure 1C). Using the Ensembl variant effect predictor (https://asia.ensembl.org/Homo_sapiens/Tools/VEP), we mapped the mutation VCF information to GRch38, excluding inconsistent chromosome/position and unmatched mutations, getting 77,700 variants (Figure 1D). Following the data processing illustrated in Figure 1, the benchmark dataset consists of 37,317 benign variants in 2,595 proteins and 40,383 pathogenic variants in 3,294 proteins. However, a subset of variants lacked annotations when processed with Ensembl VEP v104 [14]. Consequently, we excluded these variants along with the corresponding proteins. After aforementioned procedures, we finally obtained a large-scale non-redundant MM benchmark dataset. The final number of variants and proteins is presented in Table 1.
- We downloaded pathogenic GOF/LOF variants [8] (HGMD_based) from https://itanlab.shinyapps.io/goflof/, which contains a total of 9,619 variants. From this set, we selected single nucleotide variants occurring in protein coding regions excluded those that overlapped with variants in the benchmark dataset (resulting in 3,039 variants, Figure 1E). Subsequently, for the gene set within the GOF/LOF data, we searched each gene in gnomAD and applied the following filters to get benign variants: (1) removal of variants stored in ClinVar; (2) retained MMs with an allele frequency > 0.1%; (3) kept mutations with clinical significance set as benign or likely benign; (4) removed MMs that overlapped with the benchmark dataset. This process yielded 2,919 variants (Figure 1F). Finally, we merged the pathogenic and benign variants, removing redundant ones to construct blind test set. The statistical summary of variants and proteins in the benchmark dataset and blind test set is presented in the following table.
Table 1. A statistical summary of missense mutations and proteins in the benchmark dataset and blind test set.
| Data Type | Number of Variant | Number of Protein | ||
|---|---|---|---|---|
| Benign/likely benign | Pathogenic/likely pathogenic | Benign/likely benign | Pathogenic/likely pathogenic | |
| Benchmark dataset | 34,602 | 38,476 | 2,439 | 3,115 |
| Blind test set | 2,919 | 3,039 | 675 | 703 |
- ClinVar [1]: Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic acids research 2014, 42(D1):D980-D985. ClinVar website: https://www.ncbi.nlm.nih.gov/clinvar/
- dbSNP [2]: Sherry ST, Ward M-H, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic acids research 2001, 29(1):308-311. dbSNP website: https://www.ncbi.nlm.nih.gov/snp/
- gnomAD [3]: Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581(7809):434-443. gnomAD v2.1.1: https://gnomad.broadinstitute.org
- 1000 Genomes [4]: Consortium GP. A map of human genome variation from population scale sequencing. Nature 2010, 467(7319):1061. 1000 Genomes:http://www.1000genomes.org/
- UniProt [5]: UniProt: the Universal Protein knowledgebase in 2023. Nucleic Acids Research 2023, 51(D1):D523-D531. UniProt website: https://www.uniprot.org/
- ExAC [6]: Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536(7616):285-291.
- COSMIC [7]: Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Research 2011, 39(Database issue):D945-950. COSMIC website: https://cancer.sanger.ac.uk/cosmic
- GOF/LOF variants [8]: Bayrak CS, Stein D, Jain A, et al. Identification of discriminative gene-level and protein-level features associated with pathogenic gain-of-function and loss-of-function variants. The American Journal of Human Genetics 2021, 108(12):2301-2318. GOF/LOF database: https://itanlab.shinyapps.io/goflof/